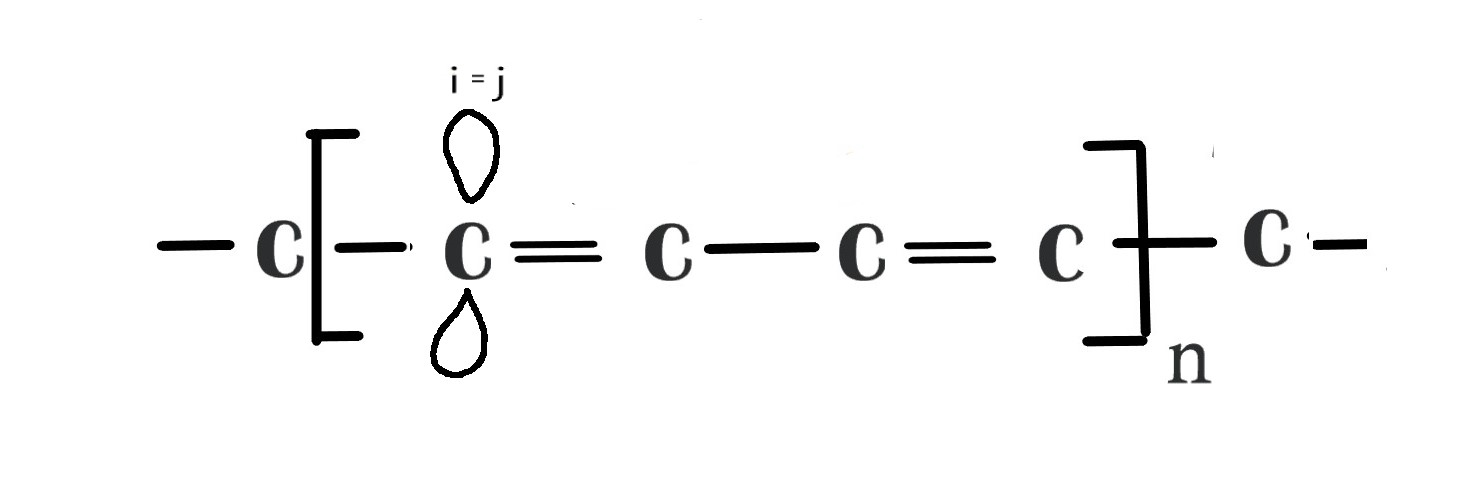Before we proceed with learning the theory, we must acquaint ourselves with certain terms related to Hückel’s rule.
Approximations – The energy of molecular orbitals is calculated by solving the Schrödinger equation for all the electrons in the molecule. However, solving the equation for molecules bigger than hydrogen is too complicated. So chemists use simplifications (approximations) that capture the main physics without doing impossible math. Thus, Approximation is a simplified model that is not 100% exact but close enough to explain or predict important behaviour.
Hückel theory makes a set ofsimplifying approximations, too. They are as follows –
Only π orbitals are considered – σ-bonds are assumed to form a stable, unchanging framework. Therefore, they are not considered when performing the calculations. Only the π electrons are considered.
Basis functions – Each carbon atom contributes exactly one 2p orbital for the π-system. Thus, only one p-orbital per carbon atom is taken into account while performing the calculations.
Overlap integrals are ignored – In reality, orbitals do overlap. However, this theory assumes that the overlap between orthogonal orbitals (orbitals oriented at right angles to each other) is zero.
Thus, the overlap integral Sij =0 for completely orthogonal orbitals i and j (no overlap).
Coulomb integrals are all the same (α) – The energy of an electron in a p-orbital on any carbon atom is taken as the same constant,α. This is the “baseline” orbital energy.
Only Resonance integrals (β) between neighbours are considered – The resonance integral (β) measures the interaction (mixing or coupling) between two orbitals on neighbouring atoms. Larger |β| means stronger interaction → stronger π-bonding and bigger splitting between bonding and antibonding MOs. The theory considers the value of β only for neighbouring carbon atoms. Thus, if two carbons are not bonded, the interaction = 0.
β is the same for all C–C bonds – Even if bonds are slightly different (like in resonance structures), Hückel takes β as a constant.
These approximations help to simplify the calculations of the Schrödinger equation. So, they are just accepted. Let us learn the approximations in detail.
Hückel’s approximations
In this theory we are going to compute two matrices namely for –
i) The overlap integral and Sij and ii) The resonance integral Hij.
i) The overlap intergral (S) – We have already studied what an overlap integral is in post # 83. Here , the overlap intergral , Sij, refers to the extent of overlap between the AOs of two pz orbitals .
If i and j are two carbon atoms under consideration, then –
If i = j i.e we are considering the AO on same carbon atom then ,Sij = 1 , as the overlap integral for an AO with itself is 1 for a normalised wavefunction. As studied in post # 83 ,normalization means integrating over the entire space of that orbital. The value 1 indicates that the overlap will be over the entire space.
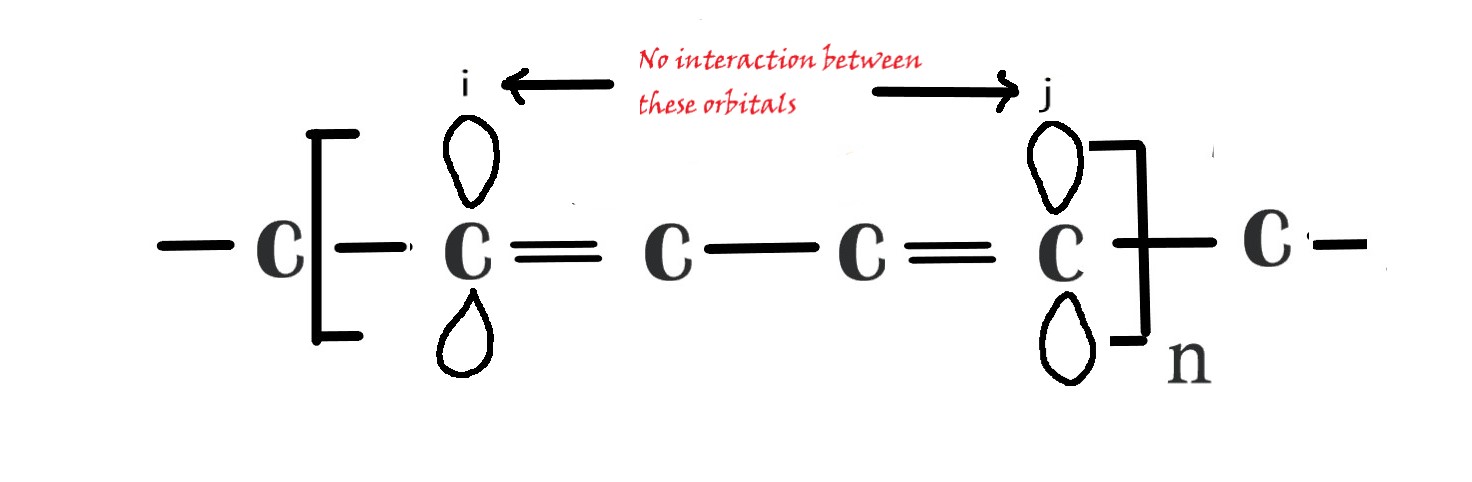
HMO theory makes an assumption ⇒ when, i≠j , then Sij = 0, which means when we are considering two AOs on different carbon atoms , the overlap integral is zero. This is because there is no interaction between these two AOs, as they are far apart and the two AOs are said to be orthogonal(do not interfere with each other).
From the above information, we can conclude that Sij is an identity matrix with all ones(1) on the diagonal and zeros(0) elsewhere. This approximation greatly reduces the complications in solving the math in HMO theory.
ii) The resonance integral, Hij – This gives us the energy of the electron in the molecular orbital. We will consider the above two cases for this integral too.
If i=j i.e we are considering only one AO (no overlap situation), Hii= α . We have already seen in post # 110 that α is the energy of 2p orbital before overlap or in no overlap situation. This explains why Hii= α.
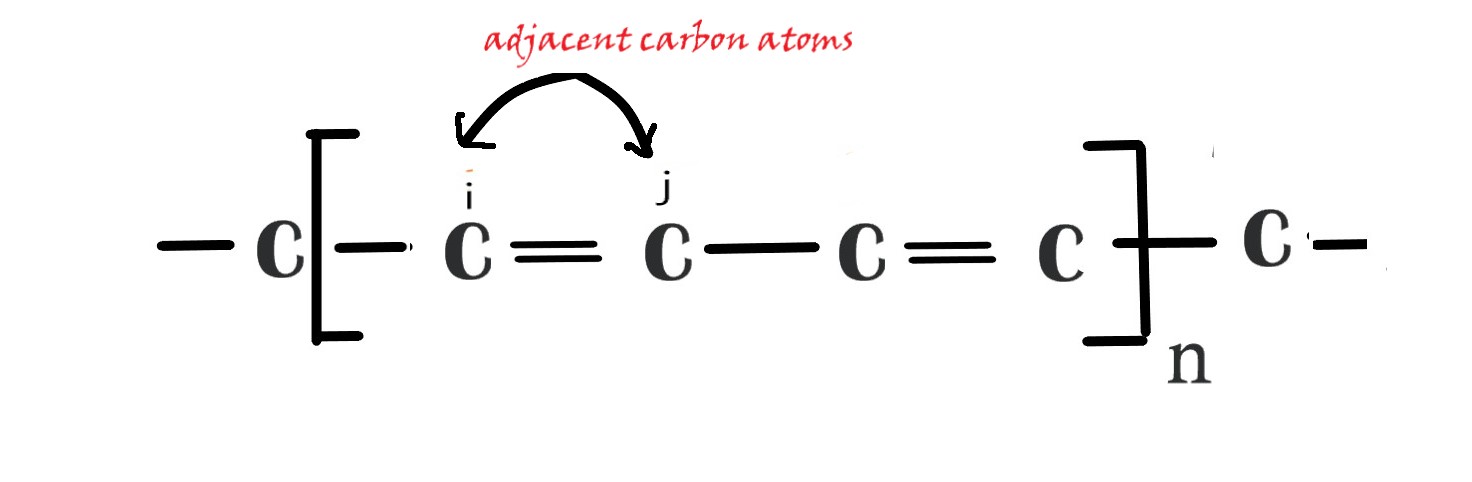
Ifi and j are adjacent carbon atoms (i= ±j e.g. -If we consider i= 2 then j has to be carbon atom #1 or 3i.e adjacent carbon) , then there will be overlap between them. We already know that ,the resonance integral (β) , gives us the energy of π bonding .So, in this case , where i and j are adjacent atoms and there is overlap, Hij= β.
If i and j are apart(two or more bonds apart) then , the resonance integral β is negligible and it is taken as zero. So, Hij= 0.
i = j
i≠j , i = j±1
i≠j , i ≠ j±1
Overlap integral Sij
1
0
0
Resonance integral Hij
α
β
0
In the next post, we will discuss more mathematical concepts required to understand the HMO theory, and then we will proceed to apply it to specific molecules.
Till then, be a perpetual student of life and keep learning…